











POTENTIAL BENEFITS OF AAV GENE THERAPY, AS A NOVEL APPROACH TO MANAGE WILSON DISEASE
Correction by gene therapy of the defective ATP7B transporter function may ultimately bring a cure to patients with Wilson disease.
The goal of gene therapy is to overcome the main limitations of current management by
– Restoring physiological copper metabolism
– Providing long-lasting benefits with a single intravenous vector administration, alleviating the need for multiple daily administration of current treatments and their associated side effects and therefore optimizing adherence to treatment
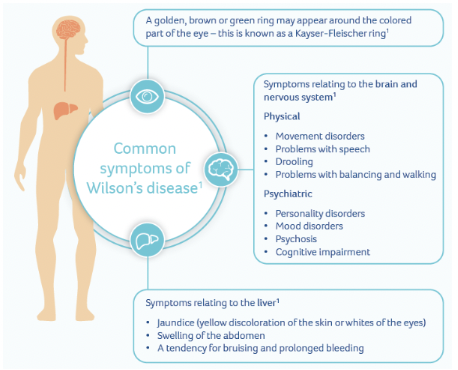
– Preventing disease complications (neurological deterioration, psychiatric manifestations and progressive liver diseases) and outweighing significant current therapy costs
Publications:
Long-term metabolic correction of Wilson’s disease in a murine model by gene therapy



A GUIDE TO GENE THERAPY
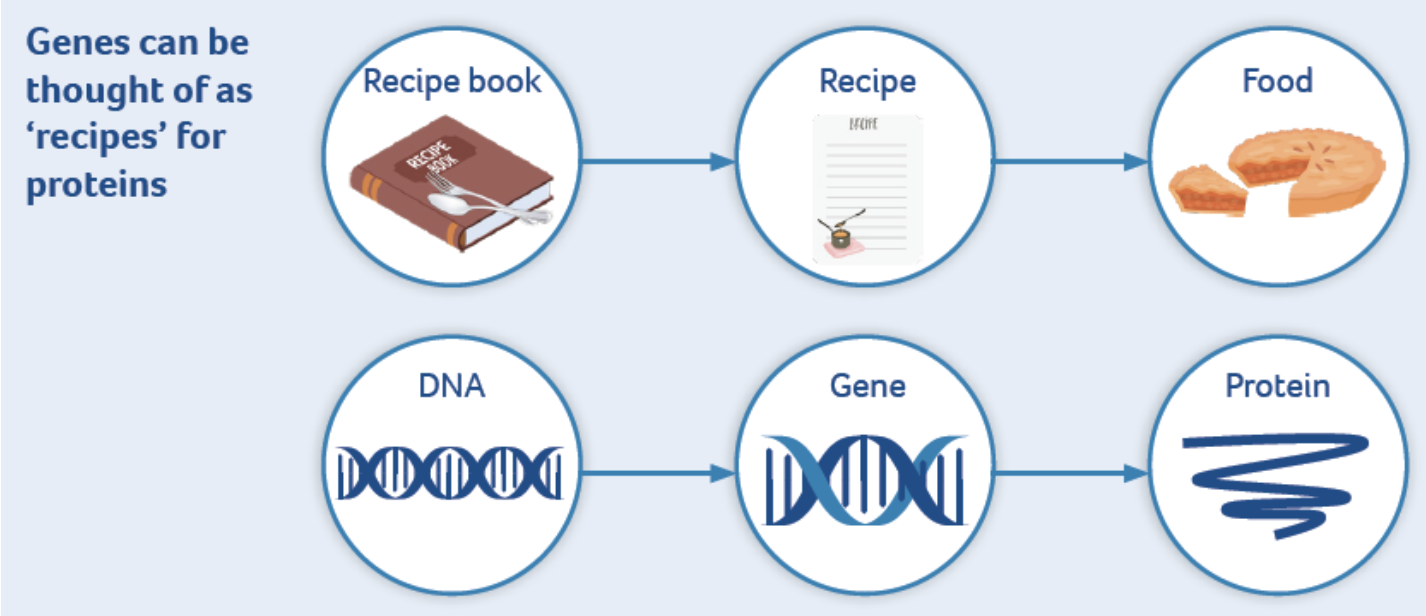
This leaflet will provide you with information about what gene therapy is, how it works and how it might help someone who has a genetic disease. This leaflet is not intended as a substitute for professional medical advice. Always seek the advice of your physician or another qualified health provider